When a rearray workflow is executed,
automatically a robot worklist suitable for executing the physical
rearraying of samples is generated. That worklist is stored, and its
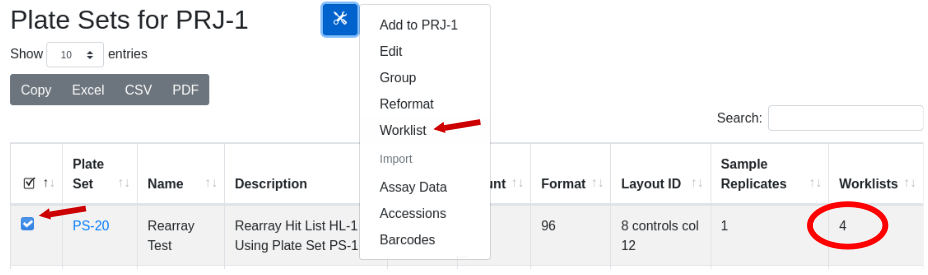
existance is indicated in the “Worklist” column in the Plate Set view.
With the plate set containing a worklist of interest selected, under the
tools icon select the menu item ‘worklist’:
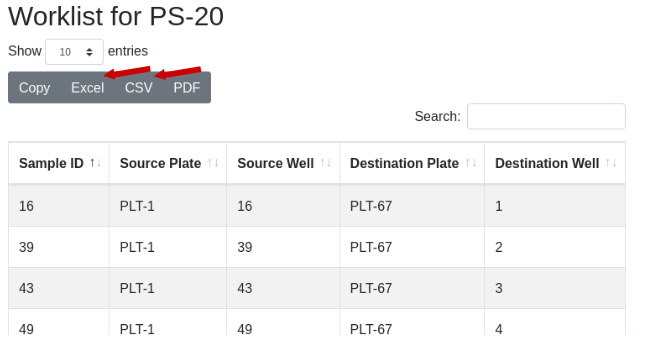
The worklist will be displayed. Use the export buttons to select the
desired export format:
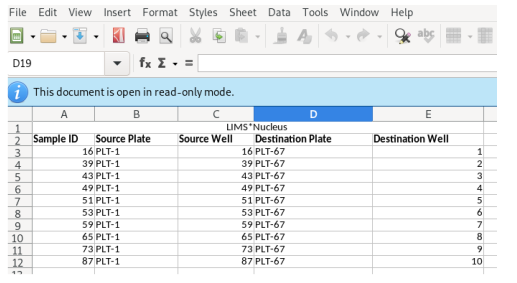
The worklist will be generated and transferred to your spreadsheet
application:
A variety of installation/configuration scripts for both the client
and the PostgreSQL database server are provided as links on this web
site or packaged with the LIMS*Nucleus client. Various scripts are
described below. Scripts without hyperlinks are included in the install
package.
Installation of the PostgreSQL database with LIMS*Nucleus tables,
methods and example data. This script is called by install-limsn-ec2.sh.
This script is only used to reinstall the database after manual
deletion
install-pg-aws-rds.sh
install database on AWS Remote Database Service PostgreSQL
instance
start-limsn.sh
Use to start the client application software. Run in detached mode
so the terminal can be shut down.
init-limsn-pack.sh
place $HOME on $PATH; modify $HOME/.bashrc; for use with Guix
pack
init-limsn-channel.sh
place $HOME on $PATH; modify $HOME/.bashrc; for use with channel
installation
load-pg.sh
load database by running all SQL scripts at command line
lnpg.sh
run lnpg.scm passing necessary parameters to initialize
database
When processing sequences obtained from a vendor, it is useful to
have an idea of how well the sequencing reactions worked, both in an
absolute sense and relative to other recently obtained sequences in the
same project. What follows is a primary sequence independent method of
evaluating a collection (i.e. and order from an outside vendor) of
sequences.
The first step is to align sequences by nucleotide index (ignoring
the actual sequence). Start by reading the sequences into a list. I use
the list s.b to hold forward (5’ to 3’) sequences, and the list s.f to
hold the reverse (but in the 5’ to 3’ orientation, as sequenced)
sequences:
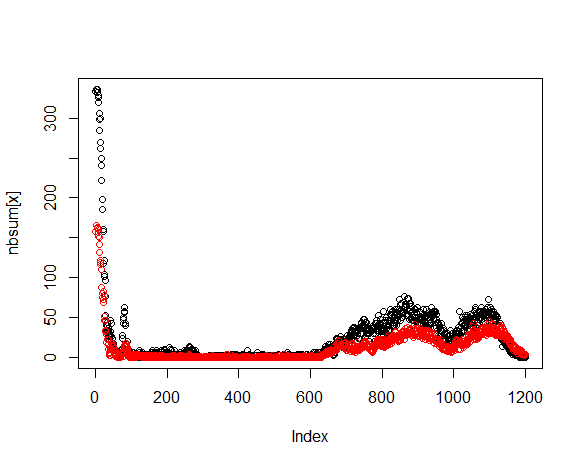
Note that ambiguities are indicated with an “n”. The sequence
evaluation will involve counting the number of ambiguitites at each
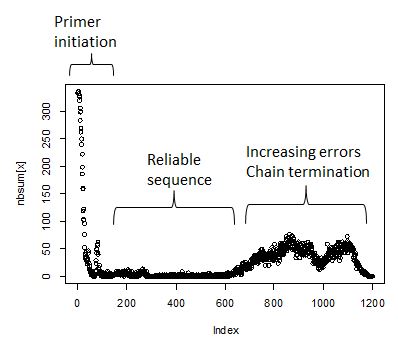
index position. The expectation is that initially - first 25 or so bases
- will have a large number of ambiguities, falling to near zero at
position 50. This is the run length required to get the primer annealed
and incoporating nucleotides. Next will follow 800-1200 positions with
near zero ambiguity count. How long exactly is a function of the
sequencing quality. Towards the end of the run the ambiguities begin to
rise as the polymerase loses energy. Finally the ambiguity count will
fall as the reads terminate.
Create a vector nbsum that will tally the count of ambiguities at a
given index. Then process through each sequence and count, at each
index, the number of ambiguities. The total count of ambiguities is
entered into nbsum at the corresponding index position.
Use R-Shiny to prototype algorithms and visualizations and extend
LIMS*Nucleus. Below is a list of assay runs from Project 1. The assay
run hyperlink transfers you to a Shiny dashboard that allows you to
manipulate and visualize your data and generate a hit list.
Default to tab delimitted text In some cases comma or tab
delimitted will be offered as an option. Proprietary formats are
avoided.
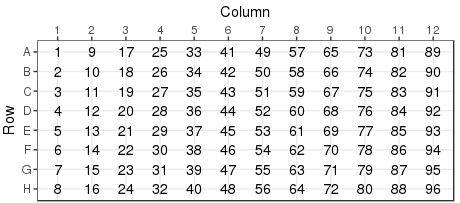
Plates are always filled by column Well number is derived from
the order of filling.
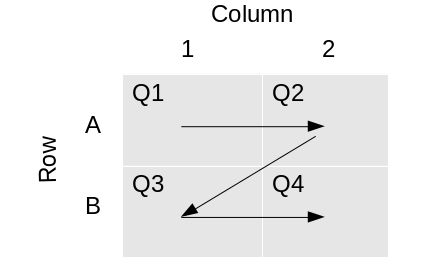
*Reformatting is performed in the “Z” pattern Quadrants are numbered
in the Z pattern.
Plate sets contain plates of the same format and layout
Always import a full plate of data, even if the plate isn’t full
e.g. a data file for three 384 well plates should have 3*384=1152 rows
even if the third plate isn’t full. Only control wells and unknown wells
with samples will be processed.
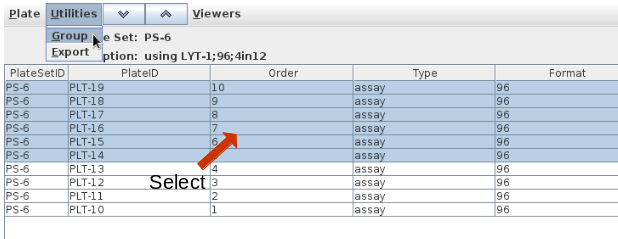
Only plate sets can be split. You can think of splitting a plate set
as a regrouping of plates within a plate set. Navigate into the plate
set of interest containing the plates to be grouped and highlight the
plates. Select Utilities/Group from the menu bar:
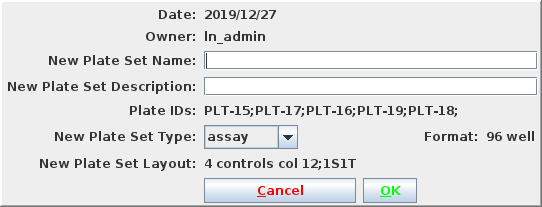
A dialog will open. Fill in the name and description for the new
plate set. The plates must be of the same format and layout, which will
be indicated in the dialog box. Select a plate type and press OK.
A systems approach involves the integration of multiple independant
commercial and custom software products to work in unison towards a
common goal. A systems approach allows flexibility by allowing for the
upgrade or discard and replacement of individual components as
requirements change.
Rob
Brigham, Amazon AWS senior manager for product management: “Now,
don’t get me wrong. It was architected in multiple tiers, and those
tiers had many components in them. But they’re all very tightly coupled
together, where they behaved like one big monolith. Now, a lot of
startups, and even projects inside of big companies, start out this way.
They take a monolith-first approach, because it’s very quick, to get
moving quickly. But over time, as that project matures, as you add more
developers on it, as it grows and the code base gets larger and the
architecture gets more complex, that monolith is going to add overhead
into your process, and that software development lifecycle is going to
begin to slow down.”
Target layouts define the pattern of targets coated on assay plates. The
available patterns are described on the replication page. Observe the patterns
under the “Target Pattern” column and note that singlicates, duplicates,
and quadruplicates are the only allowed options. Duplicates are always
in the same column, while sample duplicates are in the same row. Before
setting up a layout pattern, targets must be imported as described on
the targets page. Alternatively you can
use the built in generic targets Target1, Target2 etc. Note that
assigning targets is not required and is available only to allow merging
with target information held in other systems.
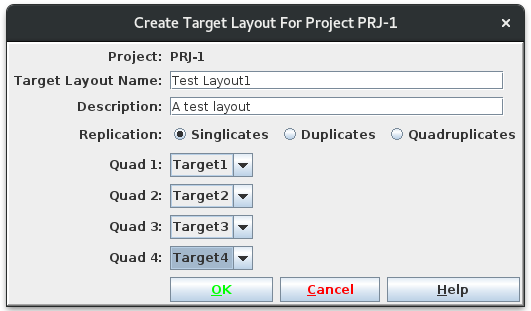
To set up a layout, navigate into the project of interest and select
the menu item Utilities/Targets/Create Target Layout. Provide a name and
description, and select the level of replication desired. The dropdowns
will be enabled as needed:
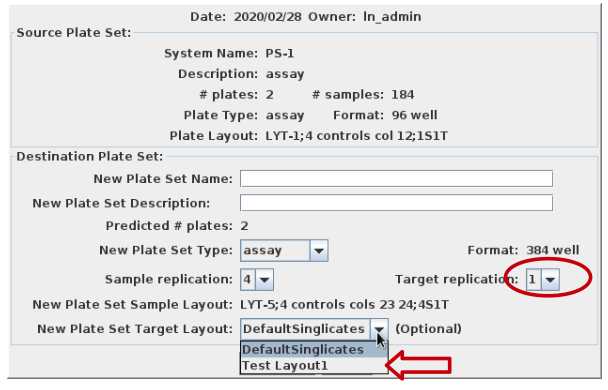
Once the layout is saved, it is available for use during reformatting
or plate set creation. Note that the layout will only appear as an
option when appropriate selections have been made e.g. replication is
singlicates:
For a definition of target see the layouts page. Targets are primarily used to
annotate data and assist with merging LIMS*Nucleus data with data from
other systems. Defining targets is optional and if not done, generic
“Target1”, “Target2” labels will be used in output. Using targets
requires three steps:
Register targets inividually or (administrator) import in bulk.
Column header spelling, capitalization, and order are critical.
Indicate the project to which the target should be associated in column
one. Import will fail if the project id is not in the database. For
targets that should be available to all projects, place “NULL” (no
quotes) in the first column. Only administrators can designate target
project id as NULL during bulk import. Note that currently there is no
opportunity to update an accession at a later time should it be blank
upon import.
One at a time import by
users
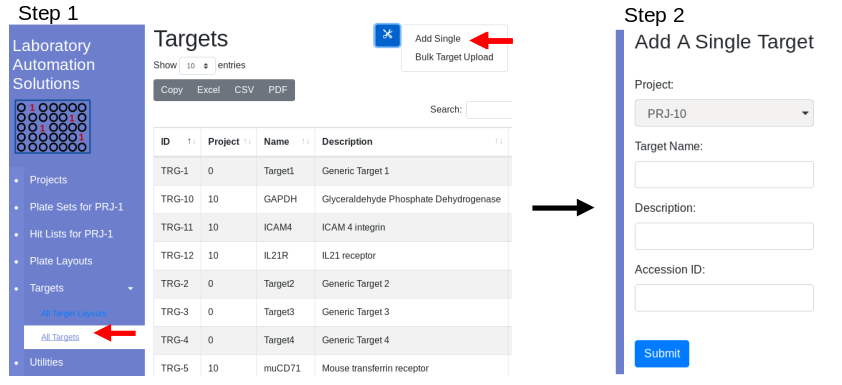
Under the menu bar Targets/Add New Target will show all targets. At
the top use the tool button to navigate to the add target page:
Fill in the form. Press Submit. The target is associated with the
current project and is only available within that project. Once targets
have been registered, they can be used in a target layout.
Sample: Item in a well.
Could be antibody, small molecule, virus, antisense oligo, expression
construct, etc. Target:
Material coated on or in an assay plate. Substance of interest that will
interact with samples. Rearray: Select random samples
(hits) across a plate and place them in a new plate. Reformat: Combine plates of one
format into a higher density plate e.g. collapse four 96 well plates
into a 384 well plate Group: Combine two or more
plate sets into one plate set; combine a subset of plates from a plate
set into a new plate set Format: Number of wells in
a plate e.g. 96, 384, 1536. Hit: A sample that
surpasses and assay threshold. Source: Plates from
which samples are drawn. Destination: Plates into
which samples are deposited. Plate set order The
assigned order of plates within a plate set. Order is visible in the
client. Required data Data required for
LIMS*Nucleus to function e.g. plate layouts, assay types, well types Example data Fake data that can be used to test
LIMS*Nucleus functionality