The QPix software identifies a single region per plate (two regions total) and randomly picks a user defined number of colonies from each region. Ideally one would want to define 96 regions per plate so that the number of colonies per region can be specified - not allowed. However each colony’s location (X|Y deck coordinate) is recorded in an XML log file.

With a bit of hacking, the well location can be assigned to each colony. In this post I will process the XML log file to extract X|Y coordinates for each picked colony, associate those coordinates with wells, associate clone IDs with wells and ultimately associate the clone ID with a picked colony.
See this post for details concerning the input files and custom methods.
1 | rm(list=ls(all=TRUE)) |
Supply plate names of the plates in which the picked colonies were deposited. This is not needed if the plate names are typed directly into the software. For this example I will assume they are not. Enter as many destination plates as exist, the script will adjust
1 |
|
Use region 2 well H01 as the “origin”. Enter calibration block values for well G01. From G01 calculate H01 using hard coded offsets.
1 |
|
Retrieve XML log files off the Qpix post run. Colony data is in //contents/colonydata. The XML can be easily visulaized by opening with a browser. Colonydata contains X|Y location as well as region. Read it in and create a dataframe.
1 | colony.data <- getNodeSet(top, "//contents/colonydata") |
Calculate centers of each column along X axis.
Offset between regions 1 and 2 is 108174um.
The custom method getDeckWell defined here
Add the source well to the dataframe.
1 |
|
Now we have associated an id based on well location to each colony.
src.well here is the well the clone came from in the transformation plate.
All we need here is colonyid assigned by Qpix and my clone id
src.well is the transformation well, keep that so you can
retrieve colonies that weren’t recovered by the Qpix.
1 |
|
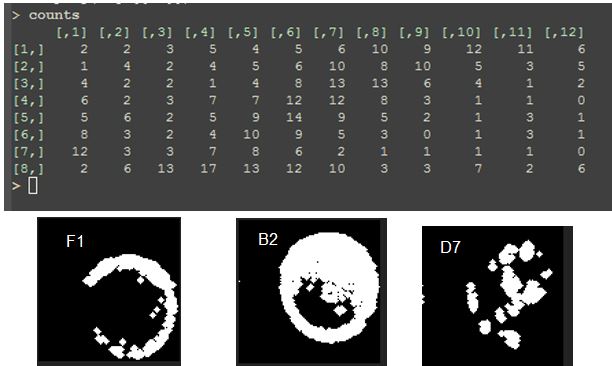
Results shows the count of colonies obtained per clone ID. Some are not present in the collection, as can be seen above where Freq==0. This provides a list of wells that can be plated manually if these are considered valuable colonies.
Pick out 4 candidates (if available) for each clone ID.
1 | d5 <- do.call("rbind", lapply( split( d4, d4$bdn ), function(x) x[1:4,]) ) |
set1 and set2 provide the plate maps for the picked plates.
results show the number of colonies picked for each candidate id. For those with 0-2 colonies, manually pick supplemental colonies if available.
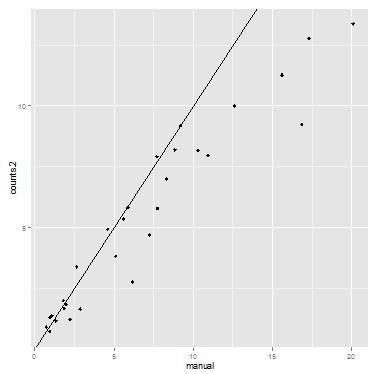
results can be plotted to get a sense for the distribution of colony counts for the various candidate clones.