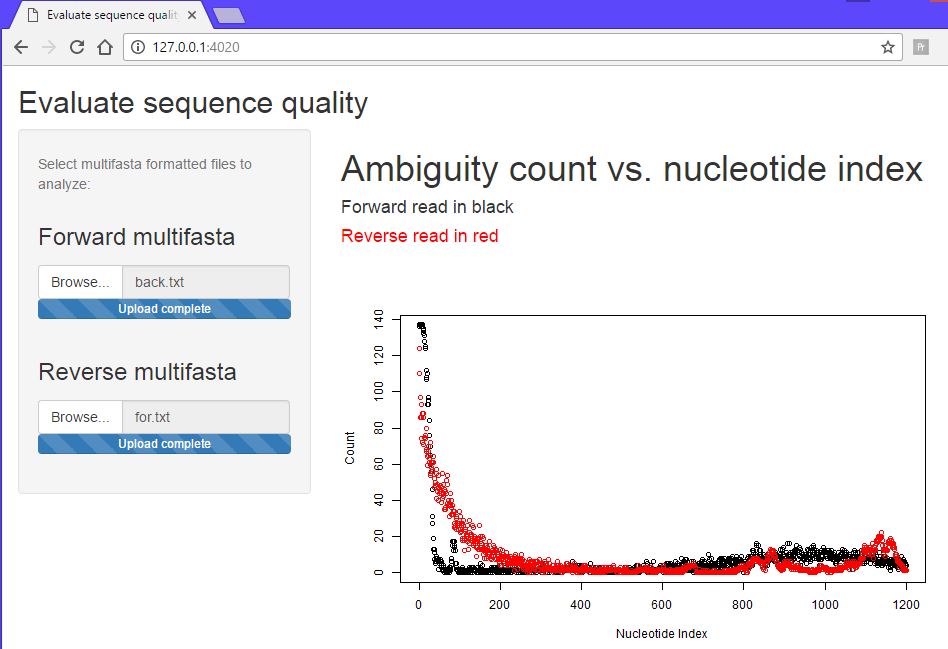
When processing sequences obtained from a vendor, it is useful to
have an idea of how well the sequencing reactions worked, both in an
absolute sense and relative to other recently obtained sequences in the
same project. What follows is a primary sequence independent method of
evaluating a collection (i.e. and order from an outside vendor) of
sequences.
The first step is to align sequences by nucleotide index (ignoring
the actual sequence). Start by reading the sequences into a list. I use
the list s.b to hold forward (5’ to 3’) sequences, and the list s.f to
hold the reverse (but in the 5’ to 3’ orientation, as sequenced)
sequences:
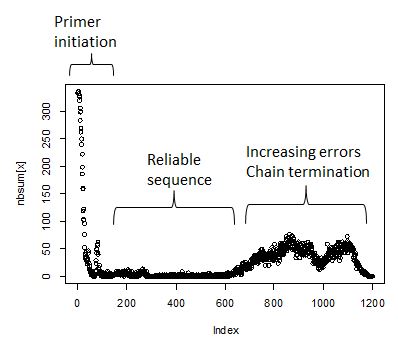
Note that ambiguities are indicated with an “n”. The sequence
evaluation will involve counting the number of ambiguitites at each
index position. The expectation is that initially - first 25 or so bases
- will have a large number of ambiguities, falling to near zero at
position 50. This is the run length required to get the primer annealed
and incoporating nucleotides. Next will follow 800-1200 positions with
near zero ambiguity count. How long exactly is a function of the
sequencing quality. Towards the end of the run the ambiguities begin to
rise as the polymerase loses energy. Finally the ambiguity count will
fall as the reads terminate.
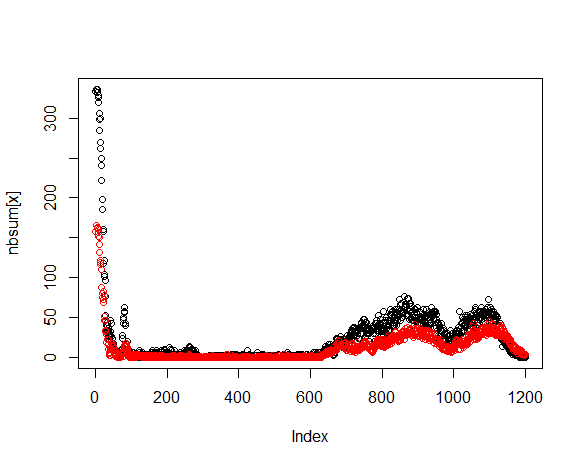
Create a vector nbsum that will tally the count of ambiguities at a
given index. Then process through each sequence and count, at each
index, the number of ambiguities. The total count of ambiguities is
entered into nbsum at the corresponding index position.