##Compare parental and mutant sequences
After perfoming error prone PCR (random) or oligonucleotide (directed) mutagenesis you will want to visualize your sequences and determine the rate of mutation incorporation. A typical visualization is the stacked bar chart as in this figure from Finlay et al. JMB (2009) 388, 541-558:
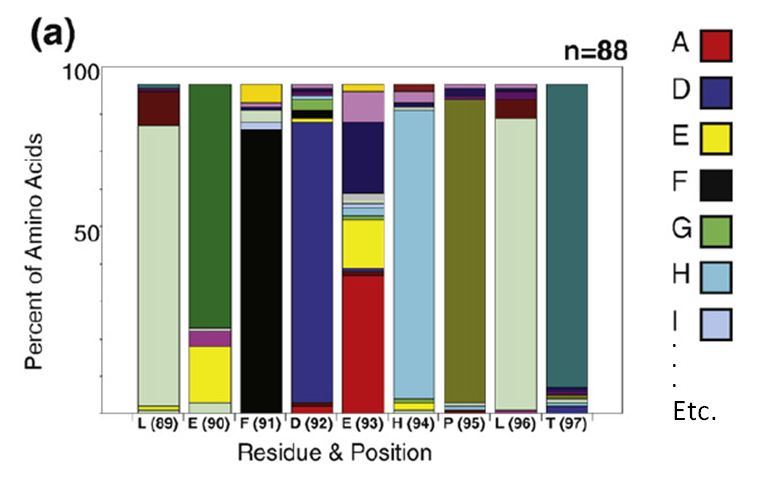
To decode this graphic you must:
- estimate the percentage of each amino acid by comparison to the Y axis
- compare relative amino acid abundance by comparing the area of boxes
- correlate color with amino acid identity
- compare to the reference sequence at the bottom of the graph
An easier to interpret graphic would be a scatter plot of sequence index (i.e. nucleotide position) on the X axis vs frequency on the Y. The data points are the single letter amino acid code. Highlight the reference sequence with a red letter.
The first step is to align all sequences. Start with a multi-fasta file of all sequences:
$cat ./myseqs.fasta
>ref
GLVQXGGSXRLSCAASGFTFSSYAMSWVRQAPGKGLEWVSAISGSGGSTYY
ADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKDHRRPKGAFDIWGQGTMVTVSS
GGGGSGGGGSGGGGSGQSALTQPASVSGSPGQSITISCTGTSSDVGAYNYVSWYQQYPGK
APKLMIYEVTNRPSGVSDRFSGSKSGNTASLTISGLQTGDEADYYCGTWDSSLSAVV
>BSA130618a-A01
glvxxggxxrlscasgftfssyamswvrqapgklewvsaisgsggstyysdsvkgrftissdnskntlylqmnslraedt
avyycakdhrrpkgafdiwgqgtmvtvssggggsggggsggggsgqsaltqprsvsgtpgqsviisctgtssdvggskyv
swyqqhpgnapkliiydvserpsgvsnrfsgsksgtsaslaitglqaedeadyycqsydsslvvf
>BSA130618a-A02
glvqpggxxrlscasgftfssyamswvrqapgkglewvsaisgsggstyyadsvkgrftisrdnskntlylqmnslraed
tavyycakdhrrpngafdiwgqgtmvtvssggggsggggsggggsgqsvvtqppsmsaapgqkvtiscsgsssnignnyv
swyqqlpgtapklliydnnkrpsxipdrfsgsksgtsatlitglqtgdeadyycgtwdsslsagvf
>BSA130618a-A03
glvqxggxxrlscasgftfssyamswvrqapgkglewvsaisgsggstyyadsvkgrftisrdnskntlylqmnslraed
tavyycakdhrrpkgafdiwgqgtmvtvssggggsggggsggggsgsyeltqppsvsvspgqtasitcsgsssniginyv
swyqqvpgtapklliyddtnrpsgisdrfsgsksgtsatlgitglqtgdeadyycgtwdsslsvvvf
Above I have labeled my parental reference sequence “ref”. Use clustalo to perform the alignment and request the output in “clustal” format. The clustalo command can be run from within R using the system command. Read the alignment file into a matrix:
input.file <- paste( getwd(), "/out.fasta", sep="")
output.file <- paste( getwd(), "/out.aln", sep="")
system( paste("c:/progra~1/clustalo/clustalo.exe -infile=", input.file, " -o ", output.file, ".aln --outfmt=clustal", sep=""))
in.file <- paste(getwd(), "/out.aln", sep="")
seqs.aln <- as.matrix(read.alignment(file = in.file, format="clustal"))
At each position determine the frequency of all 20 amino acids. Set up
a second matrix that has one dimension as the length of the sequence
and the other as 20 for each amino acid. This is the matrix that will
hold the amino acid frequencies.
The R package “seqinr” provides a constant containing all single character amino acids as well as asterisk for the stop codon. Use this to name the rows of the frequency matrix.
library(seqinr)
levels(SEQINR.UTIL$CODON.AA$L)
[1] "*" "A" "C" "D" "E" "F" "G" "H" "I" "K" "L" "M" "N" "P" "Q" "R" "S" "T" "V"
[20] "W" "Y"
aas <- c(levels(SEQINR.UTIL$CODON.AA$L), 'X')
freqs <- matrix( ncol=dim(seqs.aln)[2], nrow=length(aas))
rownames(freqs) <- aas
#Process through the matrix, calculating the frequency for each amino acid.
for( col in 1:dim(aligns)[2]){
for( row in 1:length(aas)){
freqs[row, col] <- length(which(toupper(seqs.aln[,col])==aas[row]))/dim(seqs.aln)[1]
}
}
Set up an empty plot for Frequency (Y axis) vs nucleotide index (X axis).
Y range is 0 to 1, X range is one to the length of the sequence i.e. the number of columns in the frequency matrix.
Plot frequencies >0 in black, using the single letter amino acid code as the plot character.
plot(1, type="n", xlab="Sequence Index", ylab="Frequency", xlim=c(1, dim(freqs)[2]), ylim=c(0, 1))
for( i in 1:length(aas)){
points( which(freqs[i,]>0), freqs[i, freqs[i,]>0], pch=rownames(freqs)[i], cex=0.5)
}
Overlay the reference sequence in red.
ref <-seqs.aln[rownames(seqs.aln)=="ref",]
for(i in 1:length(ref)){
if( length( freqs[rownames(freqs)[rownames(freqs)==toupper(ref[i])],i] ) > 0){
if(freqs[rownames(freqs)[rownames(freqs)==toupper(ref[i])],i] > 0){
points( i,freqs[rownames(freqs)[rownames(freqs)==toupper(ref[i])],i] , pch=toupper(ref[i]), cex=0.5, col="red")
}
}
}
This is what it looks like (open in a new tab to see detail):
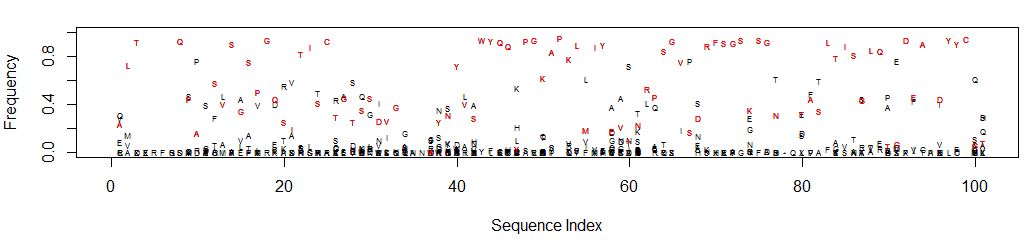
It’s easy to see which amino acid is parental, and its relative abundance to other amino acids is clear.
Consider position 61: N is the parental amino acid but T is now more abundant in the panel of mutants. K and S are the next most abundant amino acids.
Should multiple amino acids have the same or close to the same frequency, the graph can get cluttered and difficult to interpret. Adjusting the Y axis can help clarify amino acid identity. At each position percentages may not add up to 100 depending on the number of gaps. Consider the sequence “RFSGS” at positions 69-73 which is in a region containing gaps for some of the clones:
